CIDPUSA.org Autoimmune diseases
Lewis-Sumner syndrome
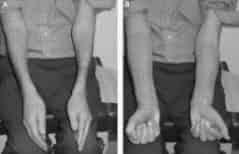
Photo shows difficulty in opening hands and difficulty in makeing a fist. Weak hands are common in Lewis Sumner syndrome and misdiagnosed as Caroal Tunnel syndrome.
Guide Book Along with autoimmune disease prevention guidelines.
Summary
The Lewis- Sumner syndrome (LSS) affects the hands causeing weakness and numbness. This is autoimmune demyelinating sensoy and motor neuropathy.
It should be considered as a clinical asymmetrical variant of chronic immune demyelinating polyneuropathy (CIDP). LSS is five times less frequent than CIDP whose prevalence is between 2 and 7/ 100,000. Patients with LSS usually present with an asymmetrical involvement of the upper limb with distal sensorimotor deficit in median or ulnar territories.
A purely sensory onset with numbness and paresthesia or pain in median or ulnar territory is observed 30% of cases. A lower limb onset is present in 30% of patients with a distal and asymmetrical sensorimotor deficit. Amyotrophy and cranial nerve involvement may be observed in 50% and 20% of patients, respectively.
LSS could mimick a nerve entrapment like carpal tunne or ulnar neuropathy or a vasculitis. The course is progressive or remitting.
Electrophysiological pattern associates a multifocal motor demyelination with conduction blocks mostly situated in the forearm.
Contrarily to CIDP, other conduction anomalies (reduction of truncal motor nerve velocities, prolonged distal latencies or prolonged F waves) occur rarely outside the blocked nerve territory.
Sensory conduction shows a multifocal sensory involvement.
Sural nerve biopsy in LSS show elements consistent with a primary demyelination, indistinguishable from that seen in typical CIDP.
However nerve biopsy is not necessary to establish the diagnosis. Serum anti-GM1 antibodies are negative and CSF protein content is usually normal or mildly elevated with a mean value of 0.7 g/l. LSS is characterized by a responsiveness to IVIg and steroids.
For LSS patients, a treatment similar to that of CIDP, with a first line treatment with intravenous Ig (IVIg) (2g/kg/course), is recommended. Patients who do not respond after 2 or 3 courses should be switched to prednisone; a dose of 1mg/kg/day should be maintained for 4-6 weeks, then slowly tapered. Plasma exchanges are not recommended in LSS. *Author: Dr K. Viala (August 2003)*.
Update : 07/08/2005
Rinsho Shinkeigaku.Clinical neurology 2000 Nov;40(11):1126-9. Lewis-Sumner syndrome presenting unilateral quadriceps amyotrophy as an initial symptom
Department of Medicine, Fukui Medical University.
We report a 55-year-old man with a chief complaint of wasting and weakness of the left quadriceps muscle. At age 54, he noticed difficulty in running and weakness in the left thigh, which
gradually progressed. On the first admission to our hospital, based on the nerve conduction studies (NCS), the muscle biopsy findings
showing neurologenic changes, and no abnormality of spinal MRI, we diagnosed as unilateral quadriceps amyotrophy, which resulted from an atypical form of spinal progressive muscular atrophy. One year
later, he showed the bilateral hand weakness, conduction blocks on the right median and ulnar nerves by NCS, and the presence of serum
anti-GM 1 antibody. From these findings, Lewis-Sumner syndrome was diagnosed. The therapy of high-dose intravenous immunoglobulin
moderately improved his symptoms. The clinical symptoms of quadriceps amyotrophy is produced by various disorders including
spinal progressive muscular atrophy, spinal extradural arachnoid cyst, rimmed vacuole myopathy, Becker dystrophy, limb-girdle
dystrophy, and focal myositis. However, there have been no reports of a case of Lewis-Sumner syndrome. It is important to consider Lewis-Sumner syndrome in the differential diagnosis of quadriceps
amyotrophy.
Publication Types:
85
B-12 deficiency
- Small Fiber neuropathy
- neurological effects of CIDP
- Nerve Fiber types
- CIDP-EMG
- Multi Focal Motor neuropathy
- M.M.F.
- Lewis Summer
- Tips for CIDP
- Axonal EMG
- CIDP-EMG
- Plasmapheresis
- CIDP-INFO
- CIDP-doctors
- CIDP-Rituxan
- CIDP-GBS-Handbook
- CIDP-family issue
- CIDP-Cyclosporin
- Polyneuropathy
- CIDP-GBS children
- Peripheral Neuropathy
- ALS & CIDP
- Types of neuropathy
- alcoholic poly neuropathy
- IVIg, Home to IVIg