pompes
Need to loose weight, recover from diabetes..
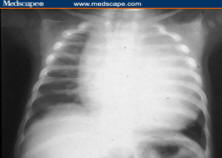
Figure 2. Severe cardiomegaly in classic infantile Pompe disease.
Progressive hypertrophic cardiomyopathy results in cardiac failure. Respiratory insufficiency and bulbar muscle weakness result in frequent respiratory infections, aspiration pneumonia, and atelectasis. Progressive respiratory insufficiency evolves in the vast majority of patients; this may be compounded by left main stem bronchus compression by a massively dilated left ventricle. Organomegaly, including hepatomegaly and macroglossia, are important clinical clues to the diagnosis. Nearly half of affected infants have feeding difficulties. Prior to the development of enzyme replacement therapy, the combination of congestive heart failure, respiratory insufficiency, and inadequate nutrition culminated in failure to thrive and early death.[4]
Children with classic infantile-onset Pompe disease may therefore have symptoms of neurologic, respiratory, cardiac, or gastrointestinal dysfunction. The physical examination is dominated by hypotonia and weakness. The symptoms of cardiomyopathy and congestive heart failure may include tachycardia and the appreciation of a cardiac murmur or gallop. The differential diagnosis of neonatal hypotonia is extensive and includes central nervous system dysfunction (hypotonic cerebral palsy, other metabolic diseases such as carnitine deficiency, mitochondrial disease, and Prader-Willi syndrome). The findings of profound hypotonia and weakness are shared with neuromuscular diseases including spinal muscular atrophy, congenital muscular dystrophies, the infantile form of myotonic dystrophy, and genetic forms of myasthenia gravis. Although the pediatrician must sequentially exclude other treatable medical causes of hypotonia, including hypothyroidism, chronic infection, malnutrition, and congenital heart disease, the finding of marked cardiomegaly in an infant with paralytic hypotonia should place Pompe disease at the top of the differential diagnosis.
Effectively Diagnosing and Treating Patients With Pompe Disease
Early Onset. The diagnosis is supported by abnormal laboratory studies including an elevated CK. Occasionally, electrodiagnostic studies (nerve conduction studies and needle electromyography) are ordered to exclude other neuromuscular diseases. The finding of widespread electrophysiologic myotonia is frequently found in infantile Pompe disease.
Early diagnosis of Pompe disease is critical for the timely introduction of supportive therapy services and genetic counseling.
Even with optimal symptomatic management, the prognosis for infantile- onset Pompe disease is grim. Prior to the onset of enzyme replacement therapy, virtually all children with this disorder died before their second birthday.[4] The natural history for these children has been dramatically altered by recombinant GAA treatments. Biweekly infusion of 20 mg/kg of recombinant GAA has prolonged survival, reversed symptomatic cardiomyopathy, and allowed a substantial number of children to achieve motor milestones unheard of in the pre-enzyme replacement era.
Late-Stage Onset. Patients with later-onset Pompe disease illustrate the clinical spectrum of this disorder. A subset of infant patients with a small amount of residual GAA activity may have a clinical course dominated by skeletal and respiratory muscle weakness in the absence of clinical or laboratory evidence of cardiomyopathy. These children have profound and life-threatening respiratory muscle insufficiency. Their management is discussed later in this article.

Links
- Home
- FAQ
- CONTACT SERVICES
- IVIG
- Healthy Oils
- Curry Powder
- Small Fiber neuropathy
- neurological effects of CIDP
- Feet Home
- Chronic fatigue syndrome
- Osteoporosis
- Women Heart Attacks
- Breast Size & Disease
- Female Sex Disease
- PARKINSON
- Memory problems
- Breast Lymph Drainage
- Kidney stone Buster
- Bras cause breast cancer
- Skin repair Clinic
- Pandas
- Hepatitis
- Weakness