Welcome to CIDPUSA.ORG
November 19, 2021
alternatives treatment of autoimmune disease read our e-book
Charcot Marie Tooth Disease
Charcot-Marie-Tooth disease (CMT) is one of the most common inherited neurological disorders, affecting approximately 1 in 2,500 people in the United States. The disease is named for the three physicians who first identified it in 1886 - Jean-Martin Charcot and Pierre Marie in Paris, France, and Howard Henry Tooth in Cambridge, England. CMT, also known as hereditary motor and sensory neuropathy (HMSN) or peroneal muscular atrophy, comprises a group of disorders that affect peripheral nerves. The peripheral nerves lie outside the brain and spinal cord and supply the muscles and sensory organs in the limbs. Disorders that affect the peripheral nerves are called peripheral neuropathies.top
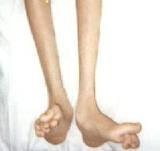 obvious muscle loss in Charcot Marie Tooth
obvious muscle loss in Charcot Marie Tooth
Professor Bob
Imperial College London
What are the symptoms of Charcot-Marie-Tooth disease?
The neuropathy of CMT affects both motor and sensory nerves. A typical feature includes weakness of the foot and lower leg muscles, which may result in foot drop and a high-stepped gait with frequent tripping or falls. Foot deformities, such as high arches and hammertoes (a condition in which the middle joint of a toe bends upwards) are also characteristic due to weakness of the small muscles in the feet. In addition, the lower legs may take on an "inverted champagne bottle" appearance due to the loss of muscle bulk. Later in the disease, weakness and muscle atrophy may occur in the hands, resulting in difficulty with fine motor skills.Onset of symptoms is most often in adolescence or early adulthood, however presentation may be delayed until mid-adulthood. The severity of symptoms is quite variable in different patients and even among family members with the disease. Progression of symptoms is gradual. Pain can range from mild to severe, and some patients may need to rely on foot or leg braces or other orthopedic devices to maintain mobility. Although in rare cases patients may have respiratory muscle weakness, CMT is not considered a fatal disease and people with most forms of CMT have a normal life expectancy.top
What are the types of Charcot-Marie-Tooth disease?
There are many forms of CMT disease, including CMT1, CMT2, CMT3, CMT4, and CMTX. CMT1, caused by abnormalities in the myelin sheath, has three main types. CMT1A is an autosomal dominant disease resulting from a duplication of the gene on chromosome 17 that carries the instructions for producing the peripheral myelin protein-22 (PMP-22). The PMP-22 protein is a critical component of the myelin sheath. An overabundance of this gene causes the structure and function of the myelin sheath to be abnormal. Patients experience weakness and atrophy of the muscles of the lower legs beginning in adolescence; later they experience hand weakness and sensory loss. Interestingly, a different neuropathy distinct from CMT1A called hereditary neuropathy with predisposition to pressure palsy (HNPP) is caused by a deletion of one of the PMP-22 genes.
Hereditary neuropathy with pressure palsies (HNPP) is an inherited condition that causes numbness, tingling and muscle weakness in the limbs. It affects the peripheral nerves, which connect your brain and spinal cord to your muscles and cells that detect touch, pain and temperature. HNPP can affect both men and women In this case, abnormally low levels of the PMP-22 gene result in episodic, recurrent demyelinating neuropathy.CMT1B is an autosomal dominant disease caused by mutations in the gene that carries the instructions for manufacturing the myelin protein zero (P0), which is another critical component of the myelin sheath. Most of these mutations are point mutations, meaning a mistake occurs in only one letter of the DNA genetic code. To date, scientists have identified more than 30 different point mutations in the P0 gene. As a result of abnormalities in P0, CMT1B produces symptoms similar to those found in CMT1A. The gene defect that causes CMT1C, which also has symptoms similar to those found in CMT1A, has not yet been identified.
CMT2 results from abnormalities in the axon of the peripheral nerve cell rather than the myelin sheath. There are many subtypes of CMT2, designated by the letters from A-L. Each subtype is characterized by the mode of inheritance and associated clinical features. The genetic loci have been identified for some subtypes. Recently, a mutation was identified in the gene that codes for the kinesin family member 1B-beta protein in families with CMT2A. Kinesins are proteins that act as motors to help power the transport of materials along the train tracks (microtubules) of the cell. Another recent finding is a mutation in the neurofilament-light gene, identified in a Russian family with CMT2E. Neurofilaments are structural proteins that help maintain the normal shape of a cell.
CMT3 or Dejerine-Sottas disease is a severe demyelinating neuropathy that begins in infancy. Infants have severe muscle atrophy, weakness, and sensory problems. This rare disorder can be caused by a specific point mutation in the P0 gene or a point mutation in the PMP-22 gene. Hypertrophic neuropathy of Dejerine-Sottas (Dejerine-Sottas syndrome ) is a term sometimes used to describe a severe, early childhood form of Charcot-Marie-Tooth disease (sometimes called type 3) that is characterized by sensory loss with ataxia in the limbs furthest from the body and pes cavus with progression towards the body.
CMT4 comprises several different subtypes of autosomal recessive demyelinating motor and sensory neuropathies. Each neuropathy subtype is caused by a different genetic mutation, may affect a particular ethnic population, and produces distinct physiologic or clinical characteristics. Patients with CMT4 generally develop symptoms of leg weakness in childhood and by adolescence they may not be able to walk. The gene abnormalities responsible for CMT4 have yet to be identified.
CMTX is an X-linked dominant disease and is caused mutation in the connexin-32 gene on the X chromosome. The connexin-32 protein is expressed in Schwann cells-cells that wrap around nerve axons, making up a single segment of the myelin sheath. This protein may be involved in Schwann cell communication with the axon. Males who inherit one mutated gene from their mothers show moderate to severe symptoms of the disease beginning in late childhood or adolescence (the Y chromosome that males inherit from their fathers does not have the connexin-32 gene). Females who inherit one mutated gene from one parent and one normal gene from the other parent may develop mild symptoms in adolescence or later or may not develop symptoms of the disease at all.