The clinician is
advised to think of
late-onset Pompe
disease as a limb
girdle syndrome with
early respiratory
involvement
(especially
orthopnea and
nocturnal
hypoventilation).
Failure to consider
this diagnosis is
all too frequent;
most patients are
symptomatic for more
than 5 years before
the diagnosis of
late-onset Pompe
disease is made.
Symptomatic
respiratory weakness
usually occurs years
after limb girdle
weakness is
established, but
patients may have
respiratory symptoms
as their initial
complaint. Muscle
biopsy is usually
helpful in
confirming the
diagnosis of
late-onset Pompe
disease. Indeed, the
muscle biopsy may
reveal the classic
changes of Pompe
disease in patients
in whom the disease
was never clinically
suspected. The
classic muscle
biopsy findings are
those of non-rimmed
vacuoles in muscle
fibers caused by
intra-lysosomal
glycogen storage.
There is increased
deposition of PAS
positive material
(Figure 5). The
vacuoles also stain
with acid
phosphatase,
confirming that the
vacuoles are
lysosomal
structures. Free
glycogen, as well as
membrane-bound
glycogen, is defined
by electron
microscopy. In
advanced disease,
the end-stage
findings of muscle
fiber atrophy and
excessive connective
tissue are present.
It is the common
wisdom that fibrosis
indicates a stage at
which muscle is
incapable of
subsequent
regeneration.
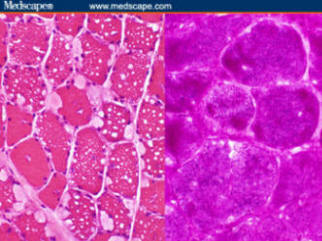
Figure 5.
Left,
hematoxylin and
eosin stain
showing
vacuolated
muscle fibers.
Right, periodic
acid-Schiff
(PAS) increased
glycogen (From
Pestronk:
Neuromuscular
Home Page)
A minority of
patients with
late-onset Pompe
disease will not
exhibit the classic
muscle biopsy
findings. Vacuolated
fibers are not found
on every biopsy.
Excess glycogen
accumulation may not
be apparent if
specific glycogen
staining with PAS is
not used. Failure to
perform muscle
ultrastructure
studies with
electron microscopy
robs the opportunity
to define the
characteristic
membrane-bound
glycogen. Even if
all studies are
performed, the
muscle biopsy is
occasionally non
diagnostic in
late-onset Pompe
disease.
The diagnostic
challenge of
late-onset Pompe
disease has
doubtlessly resulted
in failure to
diagnose a
substantial number
of these patients.
The slowly
progressive nature
of the disease leads
to compensatory
behaviors that delay
medical
consultation. In
every large
neuromuscular clinic
there are patients
with undefined limb
girdle myopathies.
The availability of
dry blood spot
technology affords a
second chance to
diagnose these
patients.[5,6]
Testing patients
with undefined limb
girdle myopathy
syndromes should
also include
pulmonary function
tests performed in
both the seated and
supine position. As
noted, a greater
than 10% drop in VC
when going from
seated to supine
indicates diaphragm
muscle weakness.
Although not
pathognemonic of
Pompe disease, the
finding of diaphragm
weakness should make
the clinician
reconsider this
diagnosis.
Late-onset Pompe
disease is an
insidiously
progressive
disorder.[11,12]
The disease has a
dramatic impact on
motor ability and
quality of life.[10]
Throughout the
course of the
disease, pelvic
girdle weakness
exceeds shoulder
girdle weakness.
The management of
both infants and
late-onset patients
requires a team
approach to
supportive care. The
services of physical
and occupational
therapists,
nutritionists,
respiratory
therapists, and
genetic counselors
play an important
role in the
management of nearly
every patient. The
patient with classic
infantile-onset
disease requires
close management by
pediatric cardiology
specialists. In
patients with
late-onset Pompe
disease, disease
duration is the
greatest predictor
of severity. Even
patients in the
early stages of
disease exhibit
diminished walking
ability and
compromised muscle
power and pulmonary
function compared
with healthy
age-matched peers.
The role of
exercise and
nutrition in Pompe
disease has been
carefully studied.
An hour a day of
treadmill walking
coupled with a
high-protein,
low-carbohydrate
diet retards disease
progression when
compliant patients
were compared with
patients who failed
to follow this
regimen.[13]
The advent of
recombinant GAA
therapy has had a
significant impact
on the longevity and
quality of life in
patients with
classic
infantile-onset
Pompe disease.
Pivotal studies
comparing these
infants with a
natural history
cohort of untreated
patients
demonstrates a
dramatic impact on
longevity, motor
development, and
somatic body growth.[7,14]
Early therapies
using GAA produced
from the milk of
transgenic rabbits
has been replaced by
rhGAA derived from
Chinese hamster
ovary cells.[8,14]
Current treatment
uses intravenous
infusion of
recombinant GAA at a
dose of 20 mg/kg on
alternate weeks. A
growing body of
evidence suggests
efficacy in those
with late-onset
disease as well.
Small pilot trials
suggest improved
power and endurance
of both limb and
respiratory muscles.
A double-blind,
controlled study
using recombinant
GAA in patients with
late-onset Pompe
disease is nearing
completion (personal
communication).
Future
therapeutic
directions will
include
second-generation
recombinant GAA.
Gene therapy with
adeno-associated
virus and other
vectors carrying the
cDNA of the GAA gene[15]
and chaperone
protein therapy to
direct native and
recombinant GAA to
the lysosome[16]
will be studied in
the future. These
treatments offer
hope for patients
and families living
with Pompe disease.
 return
to first page
return
to first page